
【深度学习模型】scMDC模型的代码复现与实验设置
论文地址:https://doi.org/10.1038/s41467-022-35031-9本文对博客中提出的scMDC模型进行复现和实验分析,scMDC 是一个端到端的深度模型,能够明确表征不同的数据源,并联合学习深度嵌入的潜在特征用于聚类分析。大量的模拟实验和真实数据实验表明,scMDC 在不同的单细胞多模态数据集上优于现有的单细胞单模态和多模态聚类方法。其运行时间的线性可扩展性使得 scM
Lin, X., Tian, T., Wei, Z. et al. Clustering of single-cell multi-omics data with a multimodal deep learning method. Nat Commun 13, 7705 (2022).
论文地址:https://doi.org/10.1038/s41467-022-35031-9
代码地址:GitHub - xianglin226/scMDC: Single Cell Multi-omics deep clustering
本文对博客论文阅读:Clustering of single-cell multi-omics data with a multimodal deep learning method-CSDN博客 z
中提出的scMDC模型进行复现和实验分析,scMDC 是一个端到端的深度模型,能够明确表征不同的数据源,并联合学习深度嵌入的潜在特征用于聚类分析。大量的模拟实验和真实数据实验表明,scMDC 在不同的单细胞多模态数据集上优于现有的单细胞单模态和多模态聚类方法。其运行时间的线性可扩展性使得 scMDC 成为分析大规模多模态数据集的有前景的方法。
目录
真实数据
一、模型代码复现
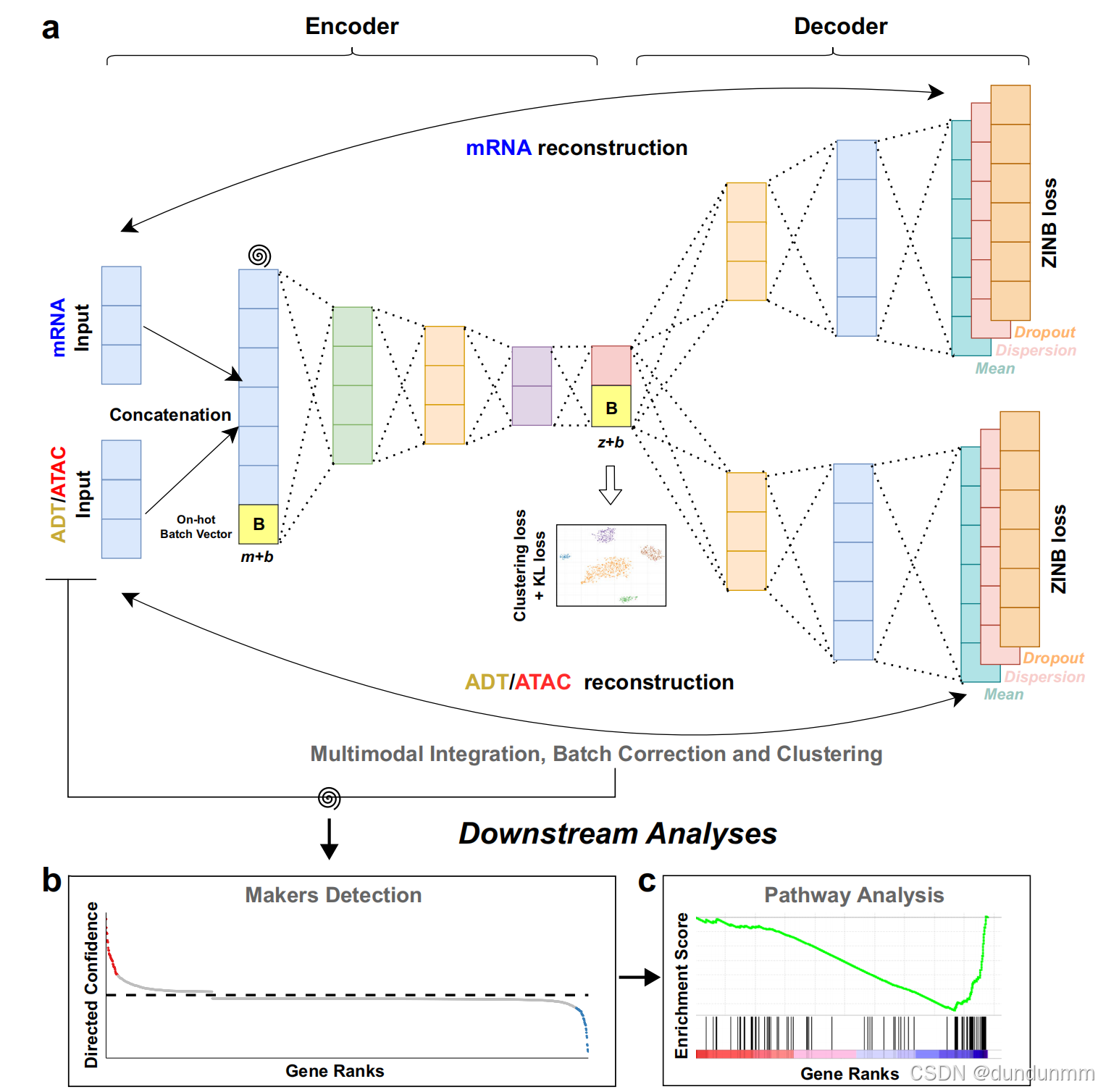
1.模型结构
scMDC 为多模态数据的连接数据设置了一个编码器,并为每种模态分别配置了一个解码器 (图 a)。它可以用于聚类 CITE-seq 数据和 10x 单细胞多组学 ATAC + 基因表达 (SMAGE-seq) 数据。螺旋符号表示人工添加的数据噪声。对于多批次数据集,scMDC 将以条件自编码器的方式运行。一个独热编码的批次向量 B(维度为 b)将分别与编码器的输入特征(原始特征维度为 m)和解码器的输入特征(潜在特征维度为 z)连接。这种设计用于批次效应校正。
scMDC 学习数据的潜在表示 Z(维度为 z),实现不同模态的整合。在 Z上实现了深度 K-means 算法和 KL损失。基于聚类结果,scMDC 利用 ACE 模型来检测不同聚类中的标志物 (图 b)。随后,可以基于 ACE 所学习的基因排序进行通路分析 (图 c)。
数据预处理
原始的CITE-seq数据通过Python包SCANPY进行预处理和标准化。mRNA和ADT数据分别进行标准化,使用相同的方法。具体来说,先过滤掉没有计数的基因和ADT。每个细胞的计数通过一个大小因子 si 进行标准化(具体来说,ADT数据使用 spi,mRNA数据使用 sri),该因子通过将该细胞的library大小除以所有细胞library大小的中位数来计算。通过这种方式,所有细胞将具有相同的library大小,从而可以进行比较。最后,计数值被转换为对数,并且被缩放至单位方差和零均值。经过处理的mRNA和ADT的计数数据用于我们的去噪多模态自编码器模型。我们使用原始计数矩阵来计算ZINB损失。
在处理单细胞多组学ATAC基因表达(SMAGE-seq)数据之前,我们将所有来自scATAC-seq的读取映射到基因区域(详细信息见下文)。然后,我们使用与CITE-seq数据相同的方法来预处理和标准化SMAGE-seq数据。ATAC数据的大小因子 sais也被计算出来。
去噪分层多模态自编码器
自编码器是一种能够高效学习非线性表示的神经网络。自编码器模型有多种类型。去噪自编码器接收带有人工噪声的损坏数据,并重构原始数据。它广泛应用于噪声数据集,以学习稳健的潜在表示。我们使用去噪自编码器处理mRNA、ADT和ATAC数据,因为它们含有大量噪声。我们用 Xr、Xp 和 Xa 来表示预处理后的mRNA、ADT和ATAC计数数据,用X_r^c、X_p^c 和X_a^c 来表示损坏的mRNA、ADT和ATAC数据,公式如下:
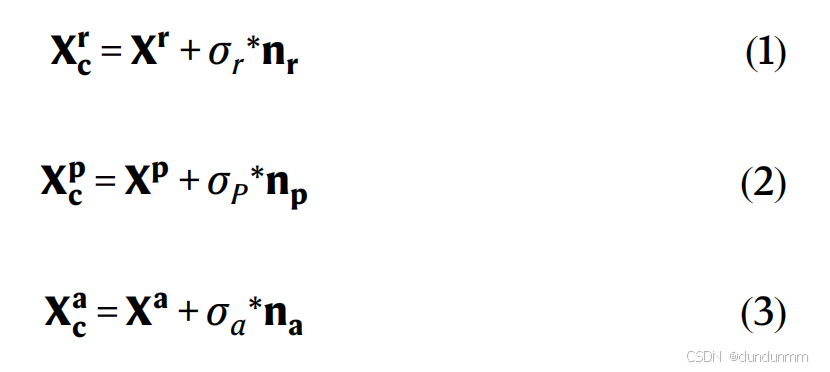
其中,nr、np 和 na 分别是mRNA、ADT和ATAC数据的人工高斯噪声(均值=0,方差=1),而 σr、σp和 σa控制着 nr、np和 na的权重。我们将 σr\设置为2.5,σp设置为1.5,σa设置为2.5。
接下来,ADT/ATAC和mRNA数据通过自编码器模型被降维到潜在空间。我们的自编码器模型包含一个用于连接数据的编码器(E),以及两个用于不同组学数据的解码器(D)。编码器和解码器都是多层全连接神经网络。我们用 Z=Ew(Xrc⊙Xpc)来表示用于连接mRNA和ADT数据的编码器,Z=Ew(Xrc⊙Xac)来表示用于连接mRNA和ATAC数据的编码器;解码器![]() 用于ATAC数据,解码器
用于ATAC数据,解码器 ![]() 用于ADT数据,解码器
用于ADT数据,解码器![]() 用于mRNA数据。Xr′、Xp′ 和 Xa′表示重建后的mRNA、ADT和ATAC数据。w和 w0分别表示编码器和解码器的可学习权重。符号 ⊙表示两个矩阵的连接操作。所有隐藏层使用ELU激活函数,且在所有隐藏层的输出上进行归一化。我们自编码器模型的重建损失函数为:
用于mRNA数据。Xr′、Xp′ 和 Xa′表示重建后的mRNA、ADT和ATAC数据。w和 w0分别表示编码器和解码器的可学习权重。符号 ⊙表示两个矩阵的连接操作。所有隐藏层使用ELU激活函数,且在所有隐藏层的输出上进行归一化。我们自编码器模型的重建损失函数为:
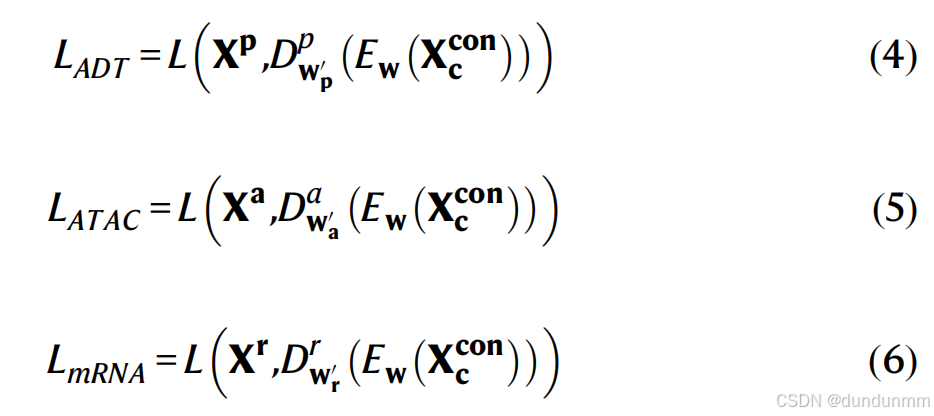
X_{con}^c 表示来自mRNA + ADT或mRNA + ATAC的数据连接。对于所有的组学数据,我们使用零膨胀负二项分布(ZINB)模型作为重建损失函数。需要注意的是,ZINB模型使用的是原始计数数据。设 X_{p_{ij}} 为原始计数矩阵ADT中细胞i和蛋白j的计数,X_{a_{ij}} 为原始计数矩阵ATAC中细胞i和基因j的计数,X_{r_{ij}} 为原始计数矩阵mRNA中细胞i和基因j的计数。负二项分布通过均值 \mu_{p_{ij}}、\mu_{a_{ij}} 和\mu_{r_{ij}} 以及离散度 \theta_{p_{ij}}、\theta_{a_{ij}} 和 \theta_{r_{ij}} 来参数化,分别对应ADT、ATAC和mRNA数据。具体公式如下:
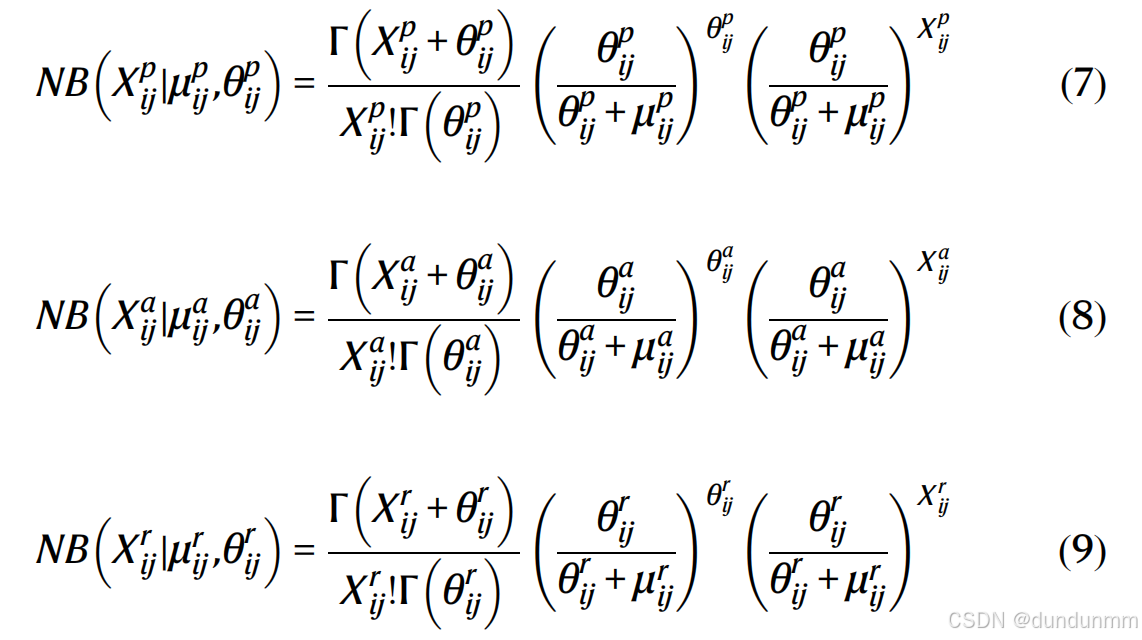
ZINB分布通过计数数据的负二项分布和一个额外的系数 \pi_{p_{ij}}、\pi_{a_{ij}} 和\pi_{r_{ij}} 来表示丢失事件的概率:
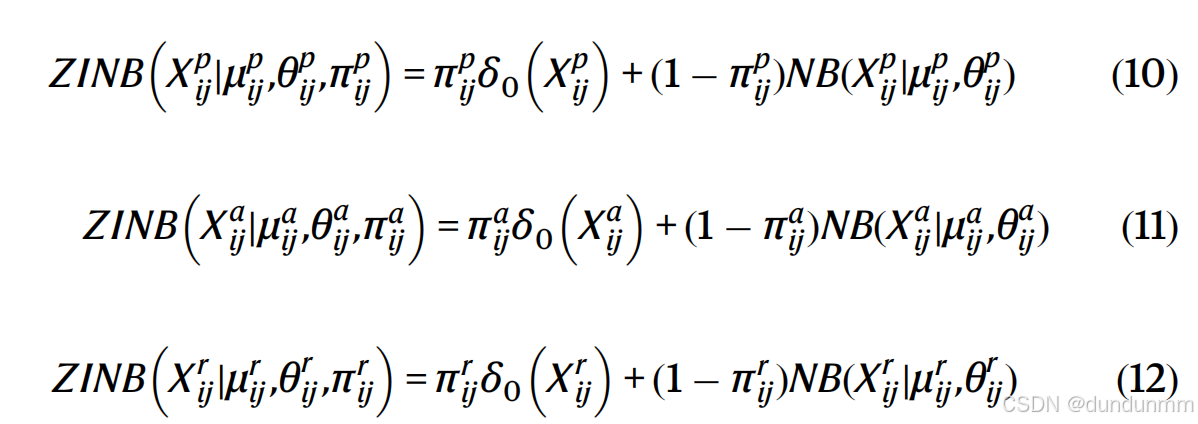
为了估计ZINB损失函数中的这些参数,我们在每个解码器的最后一个隐藏层上添加三个独立的全连接层M、θ和Π。这些层的定义如下:
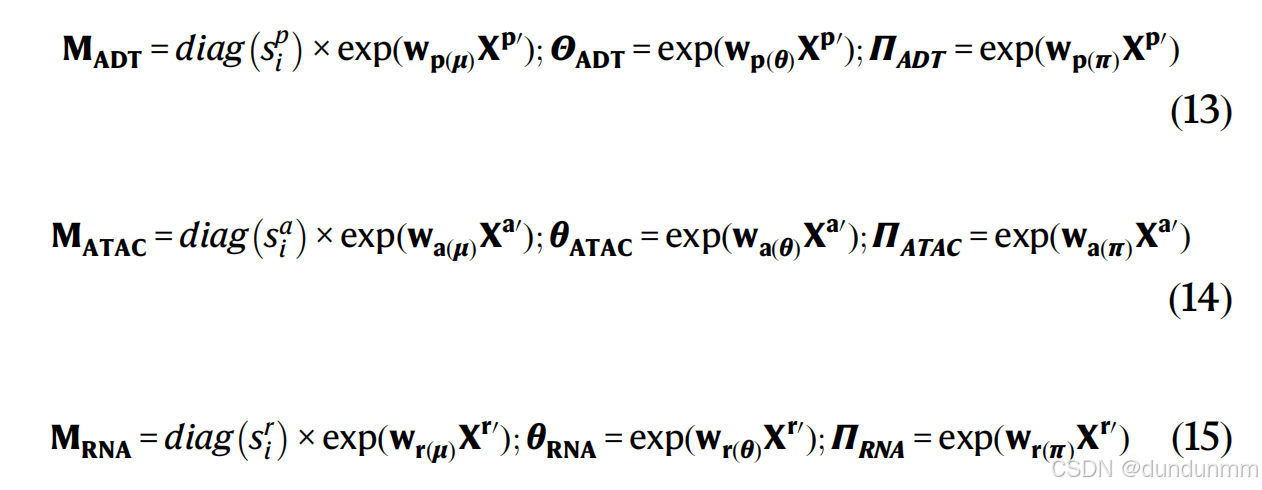
其中,M_{ADT}、\Theta_{ADT} 和\Pi_{ADT} 是用于ADT数据的ZINB损失函数的均值、离散度和丢失概率的估计矩阵,M_{ATAC}、\Theta_{ATAC} 和 Pi_{ATAC} 是用于ATAC数据的估计矩阵,M_{RNA}、\Theta_{RNA} 和 \Pi_{RNA} 是用于mRNA数据的估计矩阵。w_p(\mu)、w_p(\theta)、w_p(\pi)、w_a(\mu)、w_a(\theta)、w_a(\pi)、w_r(\mu)、w_r(\theta) 和w_r(\pi) 是可学习的权重。ADT、ATAC和mRNA的大小因子 spi、sai和 sri在预处理步骤中已经计算出来。基于ZINB的自编码器的损失函数定义为:
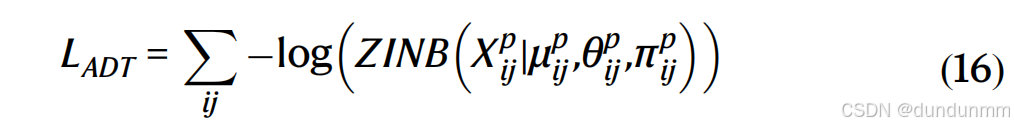
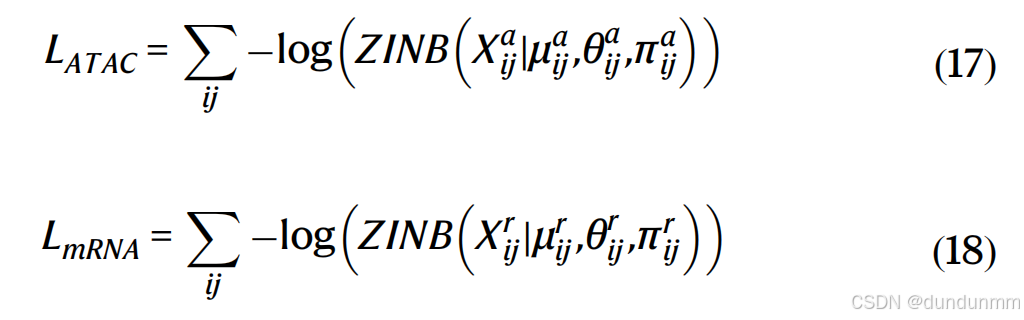
条件自编码器
条件自编码器(Conditional Autoencoder,CAE)旨在整合来自不同批次的数据。基于传统的自编码器模型,我们在编码器和解码器的输入中添加了一个矩阵B。B是来自细胞批次向量b的独热编码(one-hot encoding)。如果b中有M个批次,那么B的维度将是 N×M,其中N是细胞的数量。因此,编码器变为 Z=Ew(Xconc⊕B),而解码器变为:
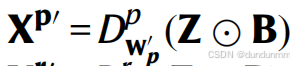 用于ADT数据,
用于ADT数据,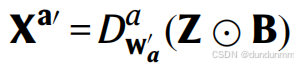 用于ATAC数据,
用于ATAC数据,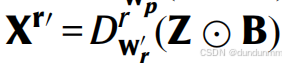 用于mRNA数据。
用于mRNA数据。
其中,⊕表示矩阵的拼接操作。
潜在层的KL散度
在聚类分析中,相似的点应该被分到同一类。根据 Chen 等人所描述的方法,我们采用 KL 散度损失函数来增强相似细胞之间的关联,并防止在潜在空间中压缩聚类的质心。继 t-SNE 之后,使用 t-分布核函数来描述我们自编码器潜在空间中两个细胞 i 和 i’ 之间的成对相似度:
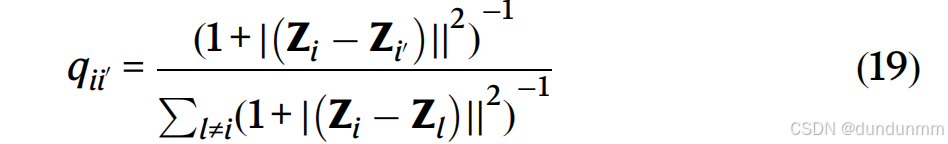
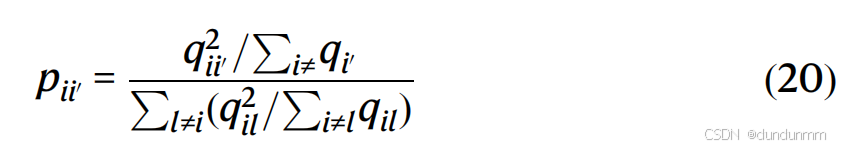
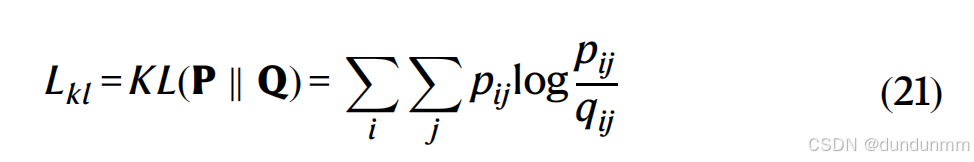
深度K-means聚类
我们在自编码器的潜在空间上执行无监督聚类。我们的多模态自编码器为每个细胞 i 学习一个非线性映射,将两个输入矩阵转换为低维空间 Z。聚类损失函数定义为:
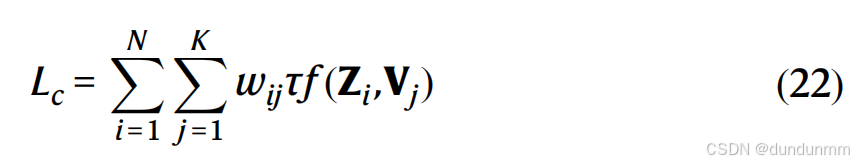
其中,V 表示 K 个聚类质心,f 计算细胞(在潜在空间中)和质心之间的欧几里得距离。τ 是一个超参数。我们为 CITE-seq 数据设置 τ 为 1,为 SMAGE-seq 数据设置 τ 为 0.1。在权重计算中应用高斯核函数,以平滑梯度下降优化过程:
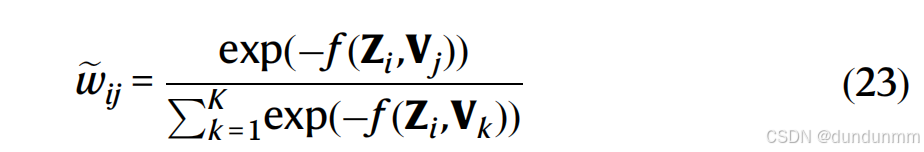
然后,为了加速收敛,对权重进行膨胀操作:

其中,超参数 α 设置为 2。
scMDC 的总损失函数定义为:
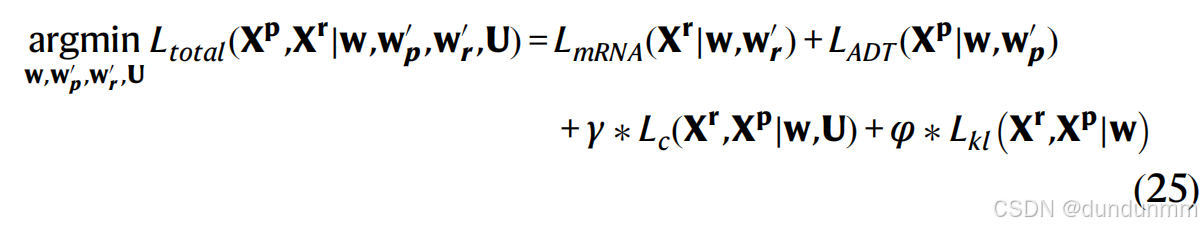
对于 CITE-seq 数据:
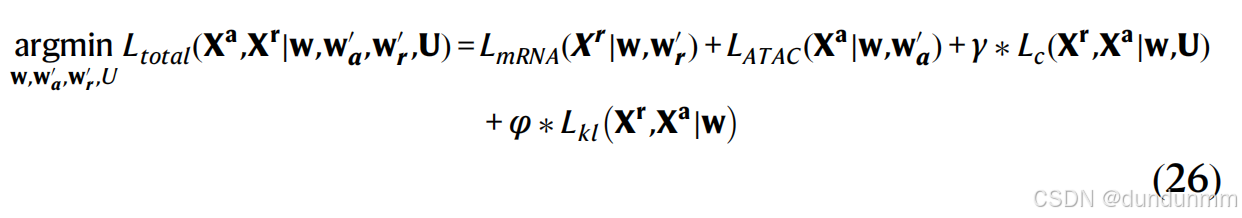
对于 SMAGE-seq 数据。w 是编码器的权重矩阵。![]() 分别是 mRNA 解码器、ADT 解码器和 ATAC 解码器的权重。U 是由 K-means 初始化的质心集合。这里,γ 和 φ 是控制聚类损失和 KL 损失权重的超参数。γ 的值在所有实验中设置为 0.1。φ 对于 CITE-seq 数据设置为 0.001,对于 SMAGE-seq 数据设置为 0.005。
分别是 mRNA 解码器、ADT 解码器和 ATAC 解码器的权重。U 是由 K-means 初始化的质心集合。这里,γ 和 φ 是控制聚类损失和 KL 损失权重的超参数。γ 的值在所有实验中设置为 0.1。φ 对于 CITE-seq 数据设置为 0.001,对于 SMAGE-seq 数据设置为 0.005。
标记基因检测
我们采用 Lu 等人提出的方法来寻找每个簇相对于另一个簇或其余簇的标记基因。简而言之,对于每个基因,该算法会找到最小的扰动,使得细胞的分组从源组 (s) 转换为目标组 (t)。一对一比较的目标函数为:
![]()
其中,折衷系数 λ 和边距 α 分别设置为 100 和 1。x ∈ X 是细胞的归一化数据,δ ∈ ℝ^P 是改变细胞聚类分配的扰动。使用 δ 的 L1 范数来鼓励稀疏性和非冗余性。一对多比较的目标函数为:
![]()
这等于将源簇与目标簇进行比较,其中细胞 x 对目标簇的置信度最高。细胞 x 对簇 c 的置信度定义为:
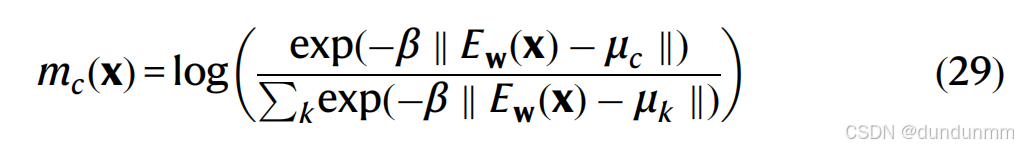
其中,μ_c 是簇 c 的质心,β 设置为 1。除了 mRNA 矩阵外,该算法还可以应用于 ADT 和 ATAC 矩阵。然后,将 ACE 学到的基因排名与基因的方向向量相乘,以得到定向的基因排名。基因的方向向量是根据簇之间的对数倍变换计算的,正值转换为 1,负值转换为 -1。基于定向基因排名,使用 R 中的 fgsea(v1.19.4)和 msigdbr(v7.4.1)包进行基因集富集分析 (GSEA)。
2.模块代码
依赖包
3.代码运行
准备输入数据,格式为 h5。(请参见“dataset”文件夹中的 readme)
根据“script”文件夹中的运行脚本运行 scMDC(如果处理的是 mRNA+ATAC 数据,并且需要进行多批次数据聚类,请注意参数设置,并使用 run_scMDC_batch.py)
根据训练好的 scMDC 模型,使用 run_LRP.py 进行差异表达分析(请参考“script”文件夹中的 LRP 运行脚本)。
二、实验分析
1.实验设置
本文中,scMDC 的所有实验均在 Nvidia Tesla P100(16G)GPU 上运行。建议在 conda 环境中安装依赖项(conda create -n scMDC)。安装依赖项仅需几分钟时间。
对于包含 5000 个细胞的数据集,scMDC 的聚类耗时约为 3 分钟。
该模型使用Python3和PyTorch实现。预训练阶段采用Adam优化器,使用AMSGrad变种,初始学习率为0.001。聚类阶段则使用Adadelta优化器,学习率为1,rho值为0.95。批次大小设置为256。在进入聚类阶段之前,我们对自编码器进行400个epoch的预训练。在预训练阶段,我们优化前200个epoch的重建损失。接下来的200个epoch中,我们在训练中加入瓶颈层的KL损失(Lkl)。预训练完成后,用户需要指定聚类的数量(K)。在聚类阶段开始时,我们通过在预训练的潜在空间上实现K-means算法来初始化K个质心。在聚类阶段,所有的损失函数,包括聚类损失(Lc),都会同时优化,质心也会在学习过程中持续更新。聚类阶段的收敛阈值是预测标签每个epoch变化小于0.1%。本研究中所有scMDC的实验都在Nvidia Tesla P100(16G)GPU上进行。
我们的模型可以用于聚类 CITE-seq 数据和 SMAGE-seq 数据。对于 CITE-seq 数据,编码器设置为 {256, 64, 32, 16},mRNA 的解码器设置为 {16, 64, 256},ADT 的解码器设置为 {16, 20}。对于 SMAGE-seq 数据,编码器设置为 {256, 128, 64},mRNA 和 ATAC 数据的解码器都设置为 {64, 128, 256}。因此,CITE-seq 和 SMAGE-seq 数据的潜在空间分别具有 16 和 64 个维度。scMDC 模型的整体架构如图 1 所示。
2.数据集
真实数据
本研究中使用的真实 CITE-seq 数据集总结在补充表 6 中。GSE100866 数据集是从 GEO 下载的(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100866)。该数据集中的细胞为脐带血单核细胞(CBMN),并由 Wang 等人根据标记基因和 ADTs 进行了注释。已过滤掉“未知”细胞类型的细胞。骨髓单核细胞(BMNC,GSE128639)及其细胞类型标签是从 SeuratData 包中的“bmcite”数据集(v0.2.1)下载的。小鼠脾脏淋巴结数据集(SLN208 和 SLN111,GSE150599)及其细胞类型标签由 TotalVI25 在 GitHub 上提供(https://github.com/YosefLab/totalVI_reproducibility)。这些细胞也经过作者过滤。PBMC 数据集可以在 10X 网站上找到(https://support.10xgenomics.com/single-cell-gene-expression/datasets)。我们从 Specter 的 GitHub 上下载了预处理数据和细胞类型标签(https://github.com/canzarlab/Specter)。
本研究中使用的真实单细胞多组学 ATAC 基因表达(SMAGE-seq)数据集总结在补充表 7 中。所有 SMAGE-seq 数据集都从 10X Genomics 网站下载(https://www.10xgenomics.com/resources/datasets)。第一个和第二个数据集来自人类外周血单核细胞(PBMCs),分别包含约 3k 和 10k 个细胞。我们分别将它们标记为 PBMC3K 和 PBMC10K。第三个数据集来自 E18 小鼠大脑,我们将其标记为 E18。对于每个数据集,mRNA 计数是直接下载的,而 ATAC 基因计数是我们生成的。具体来说,在通过 ATAC 峰区片段、核小体信号和 TSS 富集进行筛选后,我们通过 Signac(v1.4.0)62 中的“GeneActivity”功能将每个读取映射到基因区域。所有步骤都参考了 Satija 实验室的官方流程。然后,PBMC 细胞通过 Seurat V3 中的标签转移方法进行了注释,参考数据集为“pbmc_10k_v3.rds”(https://www.dropbox.com/s/zn6khirjafoyyxl/pbmc_10k_v3.rds?dl=0),由 Satija 实验室提供。对于 E18 数据集,我们从另一个小鼠大脑数据集(GSE126074 P0 小鼠大脑皮层)转移标签,细胞类型标签由 SNARE-seq 论文的作者提供。
作者在提供的代码中也提供了所有的数据集下载地址:https://drive.google.com/drive/folders/1HTCrb3HZpbN8Nte5xGhY517NMdyzqftw?usp=sharing
模拟数据
模拟数据通过 R 包 SymSim(0.0.0.9000)生成。模拟的总体设置来自 SymSim 的在线教程(https://github.com/YosefLab/SymSim/blob/master/vignettes/SymSimTutorial.Rmd)。该设置是基于 Zeisel 2015 数据集进行估算的。我们将参数“n_de_evf”降低到 5,以保持数据集中大约 50% 的差异表达基因/ADT。我们进行三个实验来测试 scMDC 的聚类性能,每个实验生成 10 个数据集。
在第一个实验中,我们调整函数 SimulateTrueCounts() 中的参数“Sigma”,将 mRNA 的值设置为 0.6、0.7 和 0.8,将 ADT 的值设置为 0.3、0.4 和 0.5,以模拟聚类中(细胞类型之间)的高、中、低聚类信号。我们给 ADT 数据设置较低的 sigma 值(较高的信号),因为在真实数据集中,ADT 数据的信噪比要高于 mRNA 数据。
在第二个实验中,我们调整函数 True2ObservedCounts() 中的参数“alpha_mean”,将 mRNA 设置为 0.001、0.00075 和 0.0005,ADT 设置为 0.05、0.045 和 0.04,以模拟低、中和高的 dropout 率。由于 mRNA 数据的 dropout 率高于 ADT 数据,这些设置与真实数据集中的观察一致。
在第三个实验中,我们在数据中添加批次效应,以测试模型在批次效应修正中的表现。此数据使用中等信号和 dropout 率,且函数 DivideBatches() 中的参数“batch_effect_size”设置为 1。
所有模拟数据集均包含 8 个组、1000 个细胞、2000 个基因和 30 个 ADT。
3.对比方法
BREM-SC(v0.2.0,GitHub地址:https://github.com/tarot0410/BREMSC)、CiteFuse(v1.0.0,GitHub地址:https://github.com/SydneyBioX/CiteFuse)、Seurat(v4.0.4,GitHub地址:https://github.com/satijalab/seurat)、IDEC(GitHub地址:https://github.com/XifengGuo/IDEC)、K-means(sklearn v0.22.2,Scikit-learn官网:https://scikit-learn.org/stable/modules/generated/sklearn.cluster.KMeans.html)、SC3(v1.21.0,GitHub地址:https://github.com/hemberg-lab/SC3)、SCVIS(v0.1.0,GitHub地址:https://github.com/shahcompbio/scvis)、Tscan(v1.31.0,GitHub地址:https://github.com/zji90/TSCAN)、TotalVI(scvi-tools v0.15.0,官网:https://scvi-tools.org/)、Cobolt(v1.0.0,GitHub地址:https://github.com/epurdom/cobolt)、scMM(v1.0.0,GitHub地址:https://github.com/kodaim1115/scMM)和Specter(GitHub地址:https://github.com/canzarlab/Specter)作为竞争方法进行比较。对于多模态方法,ADT/ATAC和mRNA数据作为输入,如果作者描述了标准化方法,则应用标准化方法。对于单一数据源方法,ADT/ATAC和mRNA矩阵分别进行预处理和标准化,然后将其连接为单一输入。为了保持一致性,所有方法使用相同的高变基因在RNA和ATAC数据中,并使用CITE-seq数据中的完整ADT数据。如果方法需要规范化数据作为输入,但未定义具体的规范化方式,则我们采用与scMDC相同的标准化方法(如上所述)。在进行K-means聚类之前,我们对标准化的mRNA数据进行PCA分析,并选择前20个主成分进行聚类。
BREM-SC直接使用原始计数矩阵作为输入。CiteFuse的标准化方法遵循该工具的vignette(https://sydneybiox.github.io/CiteFuse/articles/CiteFuse.html)。具体而言,mRNA计数通过Scater包中的“logNormCounts”函数(默认设置)进行标准化。ADT计数通过CiteFuse包中的“normalizeExprs”函数进行标准化和对数变换。Seurat使用原始计数矩阵作为输入。按照Seurat的CITE-seq教程,我们使用“LogNormalize”进行mRNA的标准化,使用“centered log-ratio transformation”进行ADT数据的标准化。然后使用“ElbowPlot”函数来查找用于聚类的最佳主成分(PC)。Seurat中的“FindClusters”函数的分辨率根据不同的数据集进行调整,以估计接近真实K值的聚类数量。对于单组学和多组学聚类,分别使用函数‘FindNeighbors’和‘FindMultiModalNeighbors’来通过SNN(共享最近邻)和WNN(加权最近邻)算法寻找细胞的邻居。
对于IDEC和Tscan,提供规范化数据作为输入。SC3需要原始数据和标准化数据作为输入。当细胞数量超过5000时,SC3会使用SVM来以监督的方式估计额外细胞的细胞类型。SCVIS是一个基于变分自编码器的模型,旨在减少scRNA-seq数据的维度。根据作者的协议(https://github.com/shahcompbio/scvis),计数数据首先处理为log2(CPM/10 + 1),其中“CPM”表示每百万计数。接着,mRNA和ADT的CPM数据连接在一起。然后通过PCA从CPM矩阵中提取前100个主成分(PC)并用作SCVIS分析的输入。K-means聚类在SCVIS的潜在输出上进行。对于TotalVI,我们根据官方管道(https://docs.scvi-tools.org/en/stable/tutorials/notebooks/totalVI.html)保持所有数据集的默认设置。我们随后在TotalVI的潜在空间上进行K-means聚类,因为假设聚类数是已知的。Specter使用标准化的RNA和ADT表达数据作为输入。我们使用Specter多模态分析的默认设置。对于SMAGE-seq数据集,我们将我们的模型与四个竞争方法进行比较:K-means + PCA、Seurat、scMM和Cobolt。所有方法使用SMAGE-seq数据中的前2000个高变mRNA和ATAC数据。如果方法需要规范化数据作为输入,我们为其应用与scMDC相同的标准化方法。在进行K-means之前,我们分别对mRNA和ATAC数据进行PCA分析,并使用前20个主成分进行聚类。对于Seurat,映射到基因区域的ATAC数据与mRNA数据以相同的方式进行处理。然后使用WNN算法来整合多模态数据,如前所述。对于Cobolt,我们遵循官方管道(https://github.com/epurdom/cobolt/blob/master/docs/tutorial.ipynb)生成数据嵌入。然后我们在数据集的潜在空间上进行K-means聚类,因为假设聚类数是已知的。我们遵循scMM(v1.0.0)提供的教程(https://github.com/kodaim1115/scMM)并使用默认参数。scMM的嵌入被获得并用于K-means聚类。
4.评价指标
调整兰德指数(ARI)、调整互信息(AMI) 和归一化互信息(NMI)被用作评估聚类性能的指标。
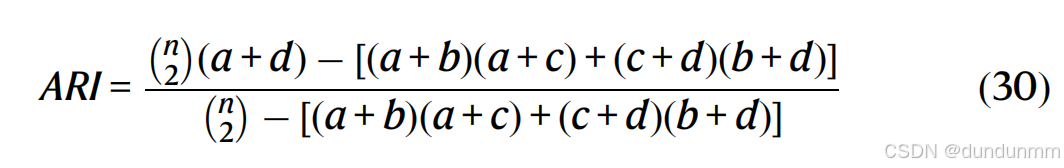
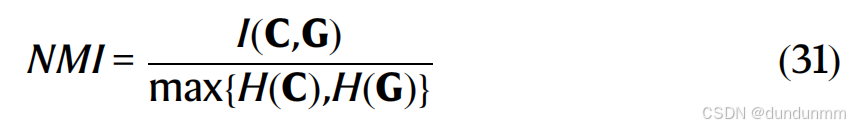
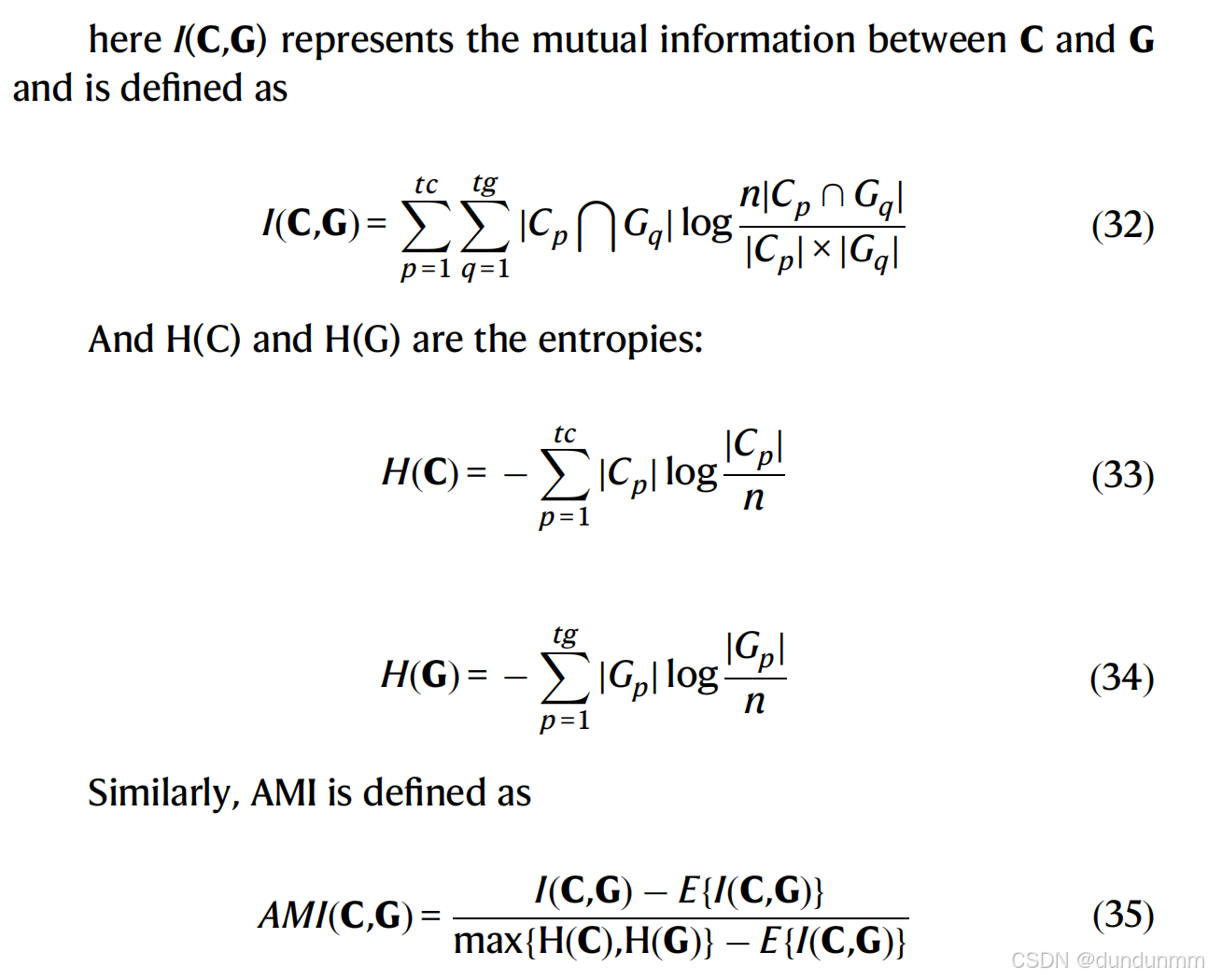
额外的成分 ![]() 是两个随机聚类之间的期望互信息。
是两个随机聚类之间的期望互信息。
为了说明 scMDC 在多个数据集上优于竞争方法,我们根据每个数据集上聚类性能(AMI、NMI 和 ARI)对方法进行排名。排名越低,性能越好。此外,我们还进行了单边配对 t 检验,以测试 scMDC 的聚类指标(NMI、AMI 和 ARI)是否显著高于竞争方法的结果,这通过 R 中的 “t.test()” 函数实现。名义 p 值 < 0.05 被认为表示存在显著差异。
模型不是很复杂,但是标记基因检测等涉及生物的知识还是理解起来很难!!
更多推荐


 34
34 0
0- 0



 已为社区贡献6条内容
已为社区贡献6条内容

所有评论(0)